喜讯 | 竺立哲教授课题组在Nature 子刊Nature Communications 发表论文
喜讯
近日,生命与健康科学学院竺立哲教授课题组与南方科技大学陈炜教授、香港科技大学黄旭辉教授和沙特阿卜杜拉国王科技大学高欣教授课题组合作,在Nature 子刊Nature Communications(《自然通讯》)上发表论文“A deep learning framework to predict binding preference of RNA constituents on protein surface”。竺立哲教授为该论文的共同通讯作者之一。
该论文提出的NucleicNet,是一种研究RBP 和RNA 结合的全新算法框架,可以同时提供 RBP 和 RNA 相互作用时的结构信息以及大规模的结合强度信息。此外,该框架可适用于其他类似问题,如蛋白质和药物小分子的相互作用,为新药研发提供新思路。
科研成果简介
RNA 结合蛋白(RBP)是基因表达调控中不可或缺的一类生物分子,对于转录后调控尤为关键。比如,Argonaute蛋白是RNA 干扰(RNAi)的核心酶, PUF 蛋白可以直接影响 mRNA 的表达等。破译RNA与蛋白相互作用的特异性和机制,对于理解RBPs功能、鉴定和识别RBPs、研究转录后调控以及设计用于RBPs识别和调节的RNAs等一系列问题都具有重要意义。
目前研究RBP 和 RNA 相互作用的实验方法可分为两类,一为基于Assay 的大规模化验分析,二为基于RBP 和RNA 结合复合物的结构分析。前者可大规模地测试 RBP 和RNA 的结合强度及RBP 对RNA 序列的选择性。基于此类实验结果的计算方法(如DeepBind等)可以整合和学习化验数据从而推断特异性模式,但无法揭示它们相互作用时的结构细节,尤其是无法鉴别其相互作用是通过碱基直接完成还是通过主链间接达成。复合物结构分析可以揭示RBP-RNA 作用细节,但受限于结构生物学实验的高昂成本无法大规模快速进行,导致在含某一特定RBP的已知RBP-RNA 复合物结构中的RNA 序列数量较少,通常不具有统计学意义。此外,已知的基于结构和序列的计算方法都只能区分结合位点和非结合位点,而无法预测RBP 对特定RNA 序列的偏好性和作用模式。
针对以上问题,该论文提出了一种仅仅基于已知的复合物结构便可预测RBP-RNA相互作用机制和特异性的深度学习算法框架NucleicNet。该方法具有以下四大功能:
(1)预测RBP与RNA具体作用模式,并将其可视化;
(2)无需大规模化验数据即可得到与实验可比的结果;
(3)可对RBP与某一RNA序列的结合强度进行评分;
(4)在不同RBPs家族中具有普适性,或可被用于识别新的RBPs及预测它们与RNA结合的位点及特异性。
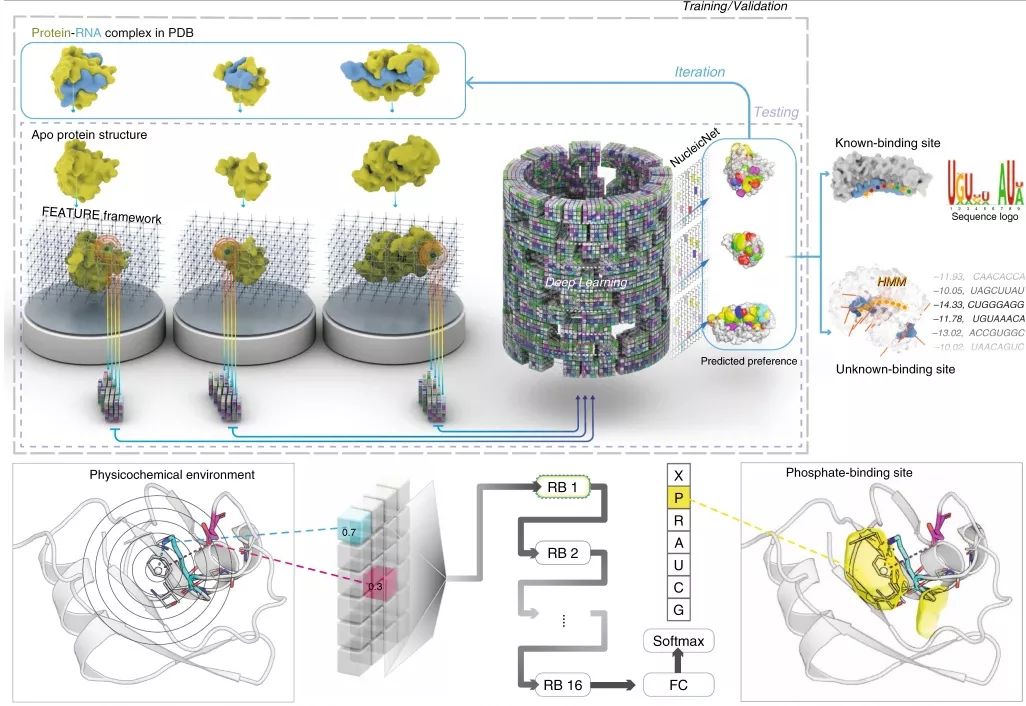
如上图所示,NucleicNet 从蛋白质的结构出发,首先在被研究蛋白质的表面产生空间点阵,然后预测空间点阵中的每一个点结合RNA 各个基团(磷酸、核糖、腺嘌呤、鸟嘌呤、胞嘧啶、尿嘧啶)的概率。对于每一个空间点,该方法使用斯坦福大学 Russ Altman 课题组(也是本文作者之一)所研发的 FEATRURE 框架去提取和该点有关的结构及理化性质信息。这些信息会被输入到一个深度学习模型之中,从而得到该点结合RNA 各个基团的概率。不同空间点与 RNA 基团的结合情况合并到一起,就可以得到RBP 与RNA 结合的结构信息,同时可预测每个点与潜在RNA序列的结合强度。由于算法本身的并行性,我们可以在短时间内预测RBP 和大量RNA 的结合机制及结合强度。对于已知的RBP,NucleicNet 可以对所有可能结合的RNA 序列进行打分和排序;对于任一蛋白质,NucleicNet 可预测其与RNA 结合的可能性(结合位点及倾向的RNA 序列),因此可用于识别该蛋白是否是RBP。
该论文同时使用了机器学习的标准评价方法以及实验方法去验证该方法的有效性。以机器学习的标准评价方式来看,该方法的准确性比其他已知方法提高了 35%。同时,在仅被用于预测RBP 上的氨基酸位点是否是RNA结合位点时,NucleicNet 的预测准确率也优于其他已知方法,尽管这并非其主要设计目标。
该论文使用了三种生物实验去验证其有效性。第一种RNACompete 实验使用RBP 在大规模的RNA 库中去竞争性地结合 RNA,从而得到该RBP 和RNA 的结合选择性。结果显示,NucleicNet 预测出的结合选择性和RNACompete 实验结果得到的结合选择性非常吻合(Pearson 相关系数高达 0.8)。
第二个实验是让NucleicNet 去区分能和 Argonaute蛋白形成成熟的RISC complex 的 guide RNA strand 以及被释放的passenger RNA strand。直观来看,guide strand 同 Argonaute 的结合性应该比passenger strand 要高,因为guide strand 最终可以和 Argonaute 结合形成了稳定的复合物。预测的结果显示,在 222 例实验中,NucleicNet 可以正确预测其中 76% 的实验,为guide strand 打出更高的结合强度评分。值得一提的是,NucleicNet 的训练数据不含有任何Assay 化验的信息,因此其预测结果与Assay 实验的高吻合度说明了NucleicNet 从结构数据中学习蛋白质和RNA 结合的统计学规律的能力。
最后一个实验是验证NucleicNet 预测的 Ago 和 siRNA 的结合强度是否和该siRNA 的敲低效率成正相关。结果显示,在 37 例基因中,22 例实验呈正相关。也就是说,尽管在活体实验条件下有众多其他因素会影响到 siRNA 的敲低效率,Argonaute 和siRNA 的结合强度仍是影响敲低效率的首要因素。这同时也意味着NucleicNet 有助于设计出敲低效率更高的siRNA。
综上所述,NucleicNet 是一种研究RBP 和RNA 结合的全新算法框架。通过使用深度学习和FEATURE 特征提取框架,该方法可以同时提供RBP 和RNA 相互作用时的结构信息以及大规模的结合强度信息。大量的计算实验和生物实验验证了该方法的有效性。同时,该框架亦或适用于其他类似问题,如蛋白质和药物小分子的相互作用,为新药研发提供了新的思路。
竺立哲教授简介

竺立哲教授是香港中文大学(深圳)生命与健康科学学院及瓦谢尔计算生物研究院的助理教授、博士生导师。他目前为本科生主讲“分子模拟与建模I”(Molecular Simulations & Modeling I),下学期将开设“计算生物学”(Computational Biology)课程。
竺立哲教授毕业于阿姆斯特丹大学化学系,曾任玛丽居里初级研究员,通过分子模拟手段研究蛋白质受体别构效应的机理。竺教授于2012年赴香港科技大学从事博士后研究,致力于增强抽样算法同马尔可夫态模型方法的整合。
竺立哲教授团队目前的研究兴趣包括RNA与蛋白质相互作用,RNA/DNA 干扰机制,高效自动化路径搜索方法,机器学习与增强采样算法的整合等。迄今已在国际主流期刊 Nature Communications,Physical Review Letters, The Journal of Physical Chemistry Letters, PLOS Computational Biology, Current Opinion in Structural Biology, Journal of Computational Chemistry 等发表论文20余篇。



